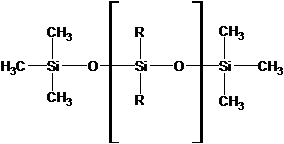Gas Chromatography TUTORIAL (GC tutorial) |
|
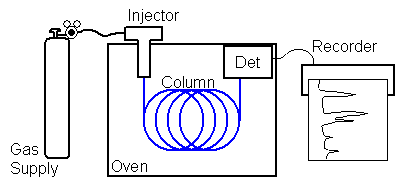
In gas chromatography (GC) the gaseous mobile phase is forced through the staionary phase using pressure. A simple GC would include a tank of gas, pressure and flow regulators to control the gas flow, an oven, an injector to allow injection of a small volume of the sample mixture under pressure, a column containing the bed of stationary phase, a detector to detect the presence of components as they exit the column, and some means to record the detector signal. A flow scheme for GC
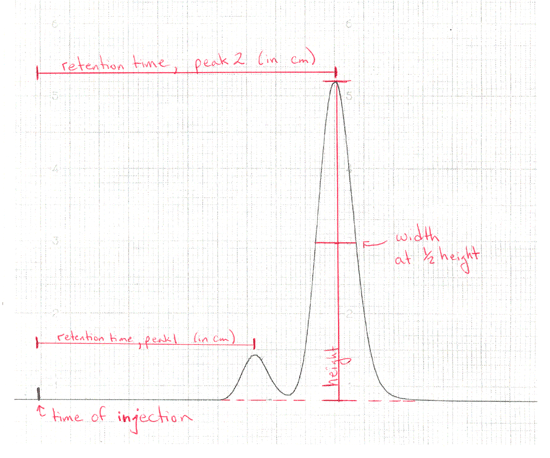
Retention timeThe time taken for a particular compound to travel through the column to the detector is known as its retention time. This time is measured from the time at which the sample is injected to the point at which the display shows a maximum peak height for that compound.Different compounds have different retention times. For a particular compound, the retention time will vary depending on:
That means that conditions have to be carefully controlled if you are using retention times as a way of identifying compounds.
Interpreting the output from the detector Report the retention time of each peak (in minutes), the identity of each component in the mixture, and the percent composition of the mixture. To determine the percent composition, you will first need to find the area under each curve.
Mark retention time, height, half-height, and width at ? height on your GC trace. |
Animation:
|
|
Theory A gas chromatograph consists of a flowing mobile phase, an injection port, a separation column containing the stationary phase, a detector, and a data recording system. The organic compounds are separated due to differences in their partitioning behavior between the mobile gas phase and the stationary phase in the column.Mobile Phase (Carrier gas) The carrier gas must be chemically inert. Commonly used gases include nitrogen, helium, argon, and carbon dioxide. The choice of carrier gas is often dependant upon the type of detector which is used. The carrier gas system also contains a molecular sieve to remove water and other impurities.
The most common stationary phases in gas-chromatography columns are polysiloxanes, which contain various substituent groups to change the polarity of the phase. The nonpolar end of the spectrum is polydimethyl siloxane, which can be made more polar by increasing the percentage of phenyl groups on the polymer. For very polar analytes, polyethylene glycol (a.k.a. carbowax) is commonly used as the stationary phase. After the polymer coats the column wall or packing material, it is often cross-linked to increase the thermal stability of the stationary phase and prevent it from gradually bleeding out of the column. Small gaseous species can be separated by gas-solid chromatography. Gas-solid chromatography uses packed columns containing high-surface-area inorganic or polymer packing. The gaseous species are separated by their size, and retention due to adsorption on the packing material.
INJECTIONPORT The sample to be analyzed is loaded at the injection port via a hypodermic syringe.The injection port is heated in order to volatilize the sample.Once in the gas phase, the sample is carried onto the column by the carrier gas, typically helium.The carrier gas is also called the mobile phase.Gas chromatographs are very sensitive instruments.Typically samples of one microliter or less are injected on the column.These volumes can be further reduced by using what is called a split injection system in which a controlled fraction of the injected sample is carried away by a gas stream before entering the column.
Detector If the column conditions are chosen correctly, the components in the sample will exit the column and flow past the detector one at a time.There are several different types of detectors common to gas chromatography instruments.The choice of detector is determined by the general class of compounds being analyzed and the sensitivity required.Flame ionization detectors (FIDs) are the most widely used detectors for organic samples.FIDs use an air/hydrogen flame to pyrolyze the effluent sample.The pyrolysis of the compounds in the flame creates ions.A voltage is applied across the flame and the resulting flow of ions is detected as a current.The number of ions produced, and therefore the resulting current, depends on the flame conditions and the identity of the molecule in question.(As a rough approximation, the current is proportional to the number of reduced carbons in the molecule.)In other words, the detector shows a different response to each compound.For this reason, separate calibrations must be performed for each compound analyzed. 1. Mass Spectrometer (GC/MS) There are two general types of column, packed and capillary (also known as open tubular). Packed columns contain a finely divided, inert, solid support material (commonly based on diatomaceous earth) coated with liquid stationary phase. Most packed columns are 1.5 - 10m in length and have an internal diameter of 2 - 4mm. Capillary columns have an internal diameter of a few tenths of a millimeter. They can be one of two types; wall-coated open tubular (WCOT) or support-coated open tubular (SCOT). Wall-coated columns consist of a capillary tube whose walls are coated with liquid stationary phase. In support-coated columns, the inner wall of the capillary is lined with a thin layer of support material such as diatomaceous earth, onto which the stationary phase has been adsorbed. SCOT columns are generally less efficient than WCOT columns. Both types of capillary column are more efficient than packed columns. In 1979, a new type of WCOT column was devised - the Fused Silica Open Tubular (FSOT) column; 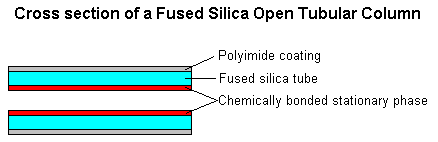
These have much thinner walls than the glass capillary columns, and are given strength by the polyimide coating. These columns are flexible and can be wound into coils. They have the advantages of physical strength, flexibility and low reactivity.
The column is where the components of the sample are separated.The column contains the stationary phase.Gas chromatography columns are of two types��packed and capillary.Capillary columns are those in which the stationary phase is coated on the interior walls of a tubular column with a small inner diameter. For example, We will use a capillary column in this experiment. The stationary phase in our column is a polysiloxane material.The basic structure of the polymeric molecules is shown below, where n indicates a variable number of repeating units and R indicates an organic functional group.In ourcolumns, 5% of the ��R��s�� are methyl groups (-CH3) and 95% of the ��R��s�� are phenyl groups (-C6H5)
This polymeric liquid has a high boiling point that prevents it from evaporating off the column during the experiment. The components in the sample get separated on the column because they take different amounts of time to travel through the column depending on how strongly they interact with the stationary phase.As the components move into the column from the injection port they dissolve in the stationary phase and are retained.Upon re-vaporization into the mobile phase they are carried further down the column.This process is repeated many times as the components migrate through the column.Components that interact more strongly with the stationary phase spend proportionally less time in the mobile phase and therefore move through the column more slowly.Normally the column is chosen such that it��s polarity matches that of the sample.When this is the case, the interaction and elution times can be rationalized according to Raoult��s law and the relationship between vapor pressure and enthalpy of vaporization.The rule of thumb is that retention times correlate with boiling points.(Do not expect an exact quantitative correlation, i.e. one with an R-value close to one, for this simple model.You will be using a non-polar column and the interaction between an alcohol molecule and the stationary phase will be dominated by weak van der Waals forces.)
What influences the separation? 1. Polarity of the stationary phasePolar compounds interact strongly with a polar stationary phase, hence have a longer retention time than non-polar columns. Chiral stationary phases based on amino acid derivatives, cyclodextrins, chiral silanes, etc are capable to separate enantiomers, because one form is slightly stronger bonded than the other one, often due to steric effects. 2. Temperature The higher the temperature, the more of the compound is in the gas phase. It does interact less with the stationary phase, hence the retention time is shorter, but the quality of separation deteriorates. 3. Carrier gas flow If the carrier gas flow is high, the molecules do not have a chance to interact with the stationary phase. The result is the same as above. 4. Column length The longer the column is the better the separation usually is. The trade-off is that the retention time increases proportionally to the column length. There is also a significant broadening of peaks observed, because of increased back diffusion inside the column. 5. Amount of material injected If too much of the sample is injected, the peaks show a significant tailing, which causes a poorer separation. Most detectors are relatively sensitive and do not need a lot of material (see below). 6. Conclusion High temperatures and high flow rates decrease the retention time, but also deteriorate the quality of the separation. ------------- Manufacturers |
| www.varianinc.com |
| www.perkinelmer.com |
| www.thermo.com |
| www.agilent.com |
| www.bdal.com |
| www.shimadzu.eu |
| www.photovac.com |
| gow-mac.com |
| www.ametekpi.com |
| www.skyrayinstrument.com |
| www.siemens.com/processautomation |
| www.abb.com |
| www.galvanic.com |
|
|
|
Cites: http://en.wikipedia.org/wiki/Gas_chromatography |
|
|