High Performance Liquid Chromatography (HPLC) TUTORIAL | ||||||||||||||||||||||||||||||||||
|
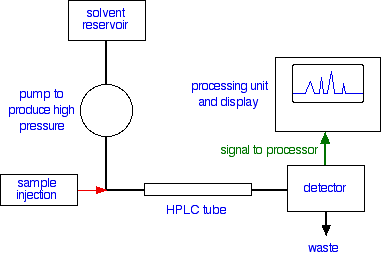
A flow scheme for HPLC
Injection of the sampleInjection of the sample is entirely automated, and you wouldn't be expected to know how this is done at this introductory level. Because of the pressures involved, it is not the same as in gas chromatography (if you have already studied that).
Retention timeThe time taken for a particular compound to travel through the column to the detector is known as its retention time. This time is measured from the time at which the sample is injected to the point at which the display shows a maximum peak height for that compound.Different compounds have different retention times. For a particular compound, the retention time will vary depending on:
That means that conditions have to be carefully controlled if you are using retention times as a way of identifying compounds.
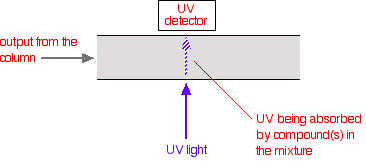
Interpreting the output from the detectorThe output will be recorded as a series of peaks - each one representing a compound in the mixture passing through the detector and absorbing UV light. As long as you were careful to control the conditions on the column, you could use the retention times to help to identify the compounds present - provided, of course, that you (or somebody else) had already measured them for pure samples of the various compounds under those identical conditions.But you can also use the peaks as a way of measuring the quantities of the compounds present. Let's suppose that you are interested in a particular compound, X.If you injected a solution containing a known amount of pure X into the machine, not only could you record its retention time, but you could also relate the amount of X to the peak that was formed.The area under the peak is proportional to the amount of X which has passed the detector, and this area can be calculated automatically by the computer linked to the display. The area it would measure is shown in green in the (very simplified) diagram.
If the solution of X was less concentrated, the area under the peak would be less - although the retention time will still be the same. For example:
This means that it is possible to calibrate the machine so that it can be used to find how much of a substance is present - even in very small quantities.Be careful, though! If you had two different substances in the mixture (X and Y) could you say anything about their relative amounts? Not if you were using UV absorption as your detection method.
In the diagram, the area under the peak for Y is less than that for X. That may be because there is less Y than X, but it could equally well be because Y absorbs UV light at the wavelength you are using less than X does. There might be large quantities of Y present, but if it only absorbed weakly, it would only give a small peak. |
||||||||||||||||||||||||||||||||||
| HPLC Simulation: Colby College Chemistry Department | ||||||||||||||||||||||||||||||||||
|
Theory Mobile Phase The mobile phase in HPLC refers to the solvent being continuously applied to the column, or stationary phase. The mobile phase acts as a carrier for the sample solution. A sample solution is injected into the mobile phase of an assay through the injector port. As a sample solution flows through a column with the mobile phase, the components of that solution migrate according to the non-covalent interactions of the compound with the column. The chemical interactions of the mobile phase and sample, with the column, determine the degree of migration and separation of components contained in the sample. For example, those samples which have stronger interactions with the mobile phase than with the stationary phase will elute from the column faster, and thus have a shorter retention time, while the reverse is also true. The mobile phase can be altered in order to manipulate the interactions of the sample and the stationary phase. There are several types of mobile phases, these include: Isocratic, gradient, and polytyptic. In isocratic elution compounds are eluted using constant mobile phase composition. The separation of compounds can be described using several equations:
In gradient elution different compounds are eluted by increasing the strength of the organic solvent. The sample is injected while a weaker mobile phase is being applied to the system. The strength of the mobile phase is later increased in increments by raising the organic solvent fraction, which subsequently results in elution of retained components. This is usually done in a stepwise or linear fashion. There are several equations that describe gradient elution: At the onset of sample introduction, the compounds are initially retained at the inlet of the column. As the solute capacity, or k', for the compound decreases, the compound begins to migrate through the stationary phase. Each of the other compounds in the sample subsequently migrate as their k' values decrease. Compared with isocratic elution, resolution and separation are improved, and bandwidths are nearly equal:? Isocratic Vs. Gradient ElutionThe Knox equation describes column efficiency or plate number N in relation to certain experimental conditions, such as column length, column diameter, temperature, flow-rate, molecular weight, etc. Plate number N is equal to plate height value H divided by particle diameter (dp). Plate height value H is in turn equal to column length L divided by N. Two of the Knox coefficients, B and C, depend on k' and size of the compound. In the equations above, k' in the isocratic equations is replaced with average k' in the gradient equations. In fact, this is the only difference in the bandwidth and resolution equations between the two. Thus, separation and height of the peak are dictated by the exact same conditions for both isocratic and gradient elution? A special feature in gradient elution is linear-solvent strength (LSS) gradients. These give approximately equal values of average k' for samples eluting at different times during separation. This is the reason why gradient elution can yield constant bandwidths for different compounds and equal resolution for pairs of compounds which have similar alpha or separation factor values. Polytyptic Mobile Phase, sometimes referred to as mixed-mode chromatography, is a versatile method in which several types of chromatographic techniques, or modes, can be employed using the same column. These columns contain rigid macroporous hydrophobic resins covalently bonded to a hydrophilic organic layer. SEC, IEC, hydrophobic or affinity chromatography are some of the methods that may be utilized. By changing the the mobile phase, the mode of separation is thereby changed which allows the chromatographer to achieve the desired selectivity in the separations. The stationary phase in HPLC refers to the solid support contained within the column over which the mobile phase continuously flows. The sample solution is injected into the mobile phase of the assay through the injector port. As the sample solution flows with the mobile phase through the stationary phase, the components of that solution will migrate according to the non-covalent interactions of the compounds with the stationary phase. The chemical interactions of the stationary phase and the sample with the mobile phase, determines the degree of migration and separation of the components contained in the sample. For example, those samples which have stronger interactions with the stationary phase than with the mobile phase will elute from the column less quickly, and thus have a longer retention time, while the reverse is also true. Columns containing various types of stationary phases are commercially available. Some of the more common stationary phases include: Liquid-Liquid, Liquid-Solid (Adsorption), Size Exclusion, Normal Phase, Reverse Phase, Ion Exchange, and Affinity. Liquid-Solid operates on the basis of polarity. Compounds that possess functional groups cabable of strong hydrogen bonding will adhere more tightly to the stationary phase than less polar compoounds. Thus, less polar compounds will elute from the column faster than compounds that are highly polar. Liquid-Liquid operates on the same basis as liquid-solid. However, this technique is better suited for samples of medium polarity that are soluble in weakly polar to polar organic solvents. The separation of non-electrolytes is achieved by matching the polarities of the sample and the stationary phase and using a mobile phase which possesses a markedly different polarity. Size-Exclusion operates on the basis of the molecular size of compounds being analyzed. The stationary phase consists of porous beads. The larger compounds will be excluded from the interior of the bead and thus will elute first. The smaller compounds will be allowed to enter the beads and will elute according to their ability to exit from the same sized pores they were internalized through. The column can be either silica or non-silica based. However, there are some size-exclusion that are weakly anionic and slightly hydrophobic which give rise to non-ideal size-exclusion behavior. Normal Phase operates on the basis of hydrophilicity and lipophilicity by using a polar stationary phase and a less polar mobile phase. Thus hydrophobic compounds elute more quickly than do hydrophilic compounds. Reverse Phase operates on the basis of hydrophilicity and lipophilicity. The stationary phase consists of silica based packings with n-alkyl chains covalently bound. For example, C-8 signifies an octyl chain and C-18 an octadecyl ligand in the matrix. The more hydrophobic the matrix on each ligand, the greater is the tendancy of the column to retain hydrophobic moieties. Thus hydrophilic compounds elute more quickly than do hydrophobic compounds. Ion-Exchange operates on the basis of selective exchange of ions in the sample with counterions in the stationary phase. IE is performed with columns containing charge-bearing functional groups attached to a polymer matrix. The functional ions are permanently bonded to the column and each has a counterion attached. The sample is retained by replacing the counterions of the stationary phase with its own ions. The sample is eluted from the column by changing the properties of the mobile phase do that the mobile phase will now displace the sample ions from the stationary phase, (ie. changing the pH). Affinity operates by using immobilized biochemicals that have a specific affinity to the compound of interest. Separation occurs as the mobile phase and sample pass over the stationary phase. The sample compound or compounds of interest are retained as the rest of the impurities and mobile phase pass through. The compounds are then eluted by changing the mobile phase conditions. Injectors Samples are injected into the HPLC via an injection port. The injection port of an HPLC commonly consists of an injection valve and the sample loop. The sample is typically dissolved in the mobile phase before injection into the sample loop. The sample is then drawn into a syringe and injected into the loop via the injection valve. A rotation of the valve rotor closes the valve and opens the loop in order to inject the sample into the stream of the mobile phase. Loop volumes can range between 10 μl to over 500 μl. In modern HPLC systems, the sample injection is typically automated. Stopped-flow Injection is a method whereby the pump is turned off allowing the injecion port to attain atmospheric pressure. The syringe containing the sample is then injected into the valve in the usual manner, and the pump is turned on. For syringe type and reciprocation pumps, flow in the column can be brought to zero and rapidly resumed by diverting the mobile phase by means of a three-way valve placed in front of the injector. This method can be used up to very high pressures? There are several types of pumps available for use with HPLC analysis, they are: Reciprocating Piston Pumps, Syringe Type Pumps, and Constant Pressure Pumps. Reciprocating Piston Pumps consist of a small motor driven piston which moves rapidly back and forth in a hydraulic chamber that may vary from 35-400 μL in volume. On the back stroke, the separation column valve is closed, and the piston pulls in solvent from the mobile phase reservoir. On the forward stroke, the pump pushes solvent out to the column from the reservoir. A wide range of flow rates can be attained by altering the piston stroke volume during each cycle, or by altering the stroke frequency. Dual and triple head pumps consist of identical piston-chamber units which operate at 180 or 120 degrees out of phase. This type of pump system is significantly smoother because one pump is filling while the other is in the delivery cycle. Syringe Type Pumps are most suitable for small bore columns because this pump delivers only a finite volume of mobile phase before it has to be refilled. These pumps have a volume between 250 to 500 mL. The pump operates by a motorized lead screw that delivers mobile phase to the column at a constant rate. The rate of solvent delivery is controlled by changing the voltage on the motor. In Constant Pressure Pumps the mobile phase is driven through the column with the use of pressure from a gas cylinder. A low-pressure gas source is needed to generate high liquid pressures. The valving arrangement allows the rapid refill of the solvent chamber whose capacity is about 70 mL. This provides continuous mobile phase flow rates. Detector The detectorThere are several ways of detecting when a substance has passed through the column. A common method which is easy to explain uses ultra-violet absorption.Many organic compounds absorb UV light of various wavelengths. If you have a beam of UV light shining through the stream of liquid coming out of the column, and a UV detector on the opposite side of the stream, you can get a direct reading of how much of the light is absorbed.The amount of light absorbed will depend on the amount of a particular compound that is passing through the beam at the time.
You might wonder why the solvents used don't absorb UV light. They do! But different compounds absorb most strongly in different parts of the UV spectrum. Methanol, for example, absorbs at wavelengths below 205 nm, and water below 190 nm. If you were using a methanol-water mixture as the solvent, you would therefore have to use a wavelength greater than 205 nm to avoid false readings from the solvent. There are various columns that are secondary to the separating column or stationary phase. They are: Guard, Derivatizing, Capillary, Fast, and Preparatory Columns. Guard Columns are placed anterior to the separating column. This serves as a protective factor that prolongs the life and usefulness of the separation column. They are dependable columns designed to filter or remove: 1) particles that clog the separation column; 2) compounds and ions that could ultimately cause "baseline drift", decreased resolution, decreased sensitivity, and create false peaks; 3) compounds that may cause precipitation upon contact with the stationary or mobile phase; and 4) compounds that might co-elute and cause extraneous peaks and interfere with detection and/or quantification. These columns must be changed on a regular basis in order to optimize their protective function. Size of the packing varies with the type of protection needed. Derivatizing Columns- Pre- or post-primary column derivatization can be an important aspect of the sample analysis. Reducing or altering the parent compound to a chemically related daughter molecule or fragment elicits potentially tangible data which may complement other results or prior analysis. In few cases, the derivatization step can serve to cause data to become questionable, which is one reason why HPLC was advantageous over gas chromatography, or GC. Because GC requires volatile, thermally stabile, or nonpolar analytes, derivatization was usually required for those samples which did not contain these properties. Acetylation, silylation, or concentrated acid hydrolysis are a few derivatization techniques. Capillary Columns- Advances in HPLC led to smaller analytical columns. Also known as microcolumns, capillary columns have a diameter much less than a millimeter and there are three types: open-tubular, partially packed, and tightly packed. They allow the user to work with nanoliter sample volumes, decreased flow rate, and decreased solvent volume usage which may lead to cost effectiveness. However, most conditions and instrumentation must be miniaturized, flow rate can be difficult to reproduce, gradient elution is not as efficient, and care must be taken when loading minute sample volumes. Fast Columns- One of the primary reasons for using these columns is to obtain improved sample throughput (amount of compound per unit time). For many columns, increasing the flow or migration rate through the stationary phase will adversely affect the resolution and separation. Therefore, fast columns are designed to decrease time of the chromatographic analysis without forsaking significant deviations in results. These columns have the same internal diameter but much shorter length than most other columns, and they are packed with smaller particles that are typically 3 μm in diameter. Advantages include increased sensitivity, decreased analysis time, decreased mobile phase usage, and increased reproducibility. Preparatory Columns- These columns are utilized when the objective is to prepare bulk (milligrams) of sample for laboratory preparatory applications. A preparatory column usually has a large column diameter which is designed to facilitate large volume injections into the HPLC system. Column efficiency refers to the performance of the stationary phase to accomplish particular separations. This entails how well the column is packed and its kinetic performance. The efficiency of a column can be measured by several methods which may or may not be affected by chromatographic anomalies, such as "tailing" or appearance of a "front." This is important because many chromatographic peaks do not appear in the preferred shape of normal Gaussian distribution. For this reason efficiency can be an enigmatic value since manufacturers may use different methods in determining the efficiency of their columns. Calculation of column efficiency value: All the following methods use this formula that measures N, or number of theoretical plates: Inflection Method- Calculation is based upon inflection point which appears at 60.7% of the peak height for a normal Gaussian peak. At this point the width of the peak is equivalent to two standard deviation units. Any asymmetrical aspect of a peak should not affect this calculation since the width is measured above the anomalous occurance (i.e., tailing or fronting). Half-peak height Method- As the name suggests, the measurement is based upon the width at 50% of peak height. For the same reason as inflection method, this measurement is not affected by asymmetry; however, this method is more reproducible from person to person since width at 50% peak height is less prone to be varied. Tangent Method- Tangent lines are drawn on each side of the peak and the width is the distance between the two lines at the base of the peak. Therefore, it is more sensitive to asymmetrical peaks and variation in efficiency values is usually seen from user to user. Sigma Methods- These methods measure peak width at decreasing levels of peak height. Thus, the three sigma method measures width at 32.4% of peak height, the four sigma method measures at 13.4%, and the five sigma method measures at 4.4%. The five sigma method is most sensitive to asymmetry because the width is measured at the lowest point. Height/Area Method- This method utilizes the fact that the area of a peak is a function of its height and standard deviation. To determine efficiency, values for peak height and area are used in a different formula: Moment Method- This method entails disregarding peak shape and expresses parameters of the peak in statistical moments. The zero moment, μ0, is the peak area. The first moment, μ1, is the mean and occurs at the center of the peak (which is the maximum peak height in normal Gaussian peaks). The second moment, μ2, is the variance of the peak. This is a detailed method where appropriate data systems are needed. For a more detailed discussion, a reference is provided. These methods were evaluated by computer simulation based on efficiency values obtained on a series of synthetically modified Gaussian peaks (i.e., increasing the 'tailing') and compared to the actual value based on the moment method (which was determined to be the most accurate). Briefly, the results were as follows: CALCULATION METHOD--ACCURACY.
| ||||||||||||||||||||||||||||||||||
| High Performance Liquid Chromatography(HPLC): A Users Guide | ||||||||||||||||||||||||||||||||||
Manufacturers
|
||||||||||||||||||||||||||||||||||
|
Cites: http://www.pharm.uky.edu/ASRG/HPLC/HPLCMYTRY.html http://en.wikipedia.org/wiki/HPLC http://www.chemguide.co.uk/analysis/chromatography/hplc.html | ||||||||||||||||||||||||||||||||||
|
|





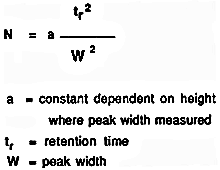 .
. 



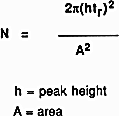 A computer is usually necessary to use this method in order to calculate the area and height.
A computer is usually necessary to use this method in order to calculate the area and height. 